Introduction
QMCPACK is an open-source, high-performance electronic structure code that implements numerous Quantum Monte Carlo (QMC) algorithms. Its main applications are electronic structure calculations of molecular, periodic 2D, and periodic 3D solid-state systems. Real-space variational Monte Carlo (VMC), diffusion Monte Carlo (DMC), and a number of other advanced QMC algorithms are implemented. A full set of orbital-space auxiliary-field QMC (AFQMC) methods is also implemented. By directly solving the Schrodinger equation, QMC methods offer greater accuracy than methods such as density functional theory but at a trade-off of much greater computational expense. Distinct from many other correlated many-body methods, QMC methods are readily applicable to both isolated molecular systems and to bulk (periodic) systems including metals and insulators. The few systematic errors in these methods are increasingly testable allowing for greater confidence in predictions and convergence to e.g. chemically accurate results in some cases.
QMCPACK is written in C++ and is designed with the modularity afforded by object-oriented and generic programming. High parallel and computational efficiencies are achievable on workstations through to the largest supercomputers. Because of the modular architecture, the addition of new wavefunctions, algorithms, and observables is relatively straightforward. For parallelization, QMCPACK uses a fully hybrid OpenMP/MPI approach to optimize memory usage and to take advantage of many-core CPU nodes. OpenMP offload and a limited amount of CUDA/HIP/SYCL is utilized for support of NVIDIA, AMD, and Intel graphical processing units (GPUs) and accelerators. Finally, QMCPACK uses standard file formats for input and output in XML and HDF5 to facilitate data exchange.
This manual currently serves as an introduction to the essential features of QMCPACK and as a guide to installing and running it. Over time this manual will be expanded to include a fuller introduction to QMC methods in general and to include more of the specialized features in QMCPACK.
Besides studying this manual we recommend reading a recent review of QMCPACK developments [KAB+20] as well as the QMCPACK citation paper [KBB+18].
Quickstart and a first QMCPACK calculation
In case you are keen to get started, this section describes how to quickly build and run a first QMCPACK calculation on a standard UNIX or Linux-like system. The build system usually works without much fuss on these systems. If C++, MPI, BLAS/LAPACK, FFTW, HDF5, and CMake are already installed, QMCPACK can be built and run within five minutes. For supercomputers, cross-compilation systems, and some computer clusters, the build system might require hints on the locations of libraries and which versions to use, typical of any code; see Obtaining, installing, and validating QMCPACK. Installation instructions for common workstations and supercomputers includes complete examples of installations for common workstations and supercomputers that you can reuse.
To build QMCPACK:
Download the latest QMCPACK distribution via https://www.qmcpack.org.
Untar the archive (e.g.,
tar xvf v4.0.0.tar.gz).Check the instructions in the README file.
Run CMake in a suitable build directory to configure QMCPACK for your system:
cd qmcpack/build; cmake ..If CMake is unable to find all needed libraries, see Obtaining, installing, and validating QMCPACK for instructions and specific build instructions for common systems.
Build QMCPACK:
makeormake -j 16; use the latter for a faster parallel build on a system using, for example, 16 processes.The QMCPACK executable is usually
bin/qmcpack. If you build the complex version it isbin/qmcpack_complex.
QMCPACK is distributed with examples illustrating different capabilities. Most of the examples are designed to run quickly with modest resources. We’ll run a short diffusion Monte Carlo calculation of a water molecule:
Go to the appropriate example directory:
cd ../examples/molecules/H2O.- (Optional) Put the QMCPACK binary on your path:
export PATH=\$PATH:location-of-qmcpack/build/bin Run QMCPACK:
../../../build/bin/qmcpack simple-H2O.xmlorqmcpack simple-H2O.xmlif you followed the step above.The run will output to the screen and generate a number of files:
$ls H2O* H2O.HF.wfs.xml H2O.s001.scalar.dat H2O.s002.cont.xml H2O.s002.qmc.xml H2O.s002.stat.h5 H2O.s001.qmc.xml H2O.s001.stat.h5 H2O.s002.dmc.dat H2O.s002.scalar.dat
Partially summarized results are in the standard text files with the suffixes scalar.dat and dmc.dat. They are viewable with any standard editor.
If you have Python and matplotlib installed, you can use the analysis utility to produce statistics and plots of the data. See Analyzing QMCPACK data for information on analyzing QMCPACK data.
export PATH=$PATH:location-of-qmcpack/nexus/bin
export PYTHONPATH=$PYTHONPATH:location-of-qmcpack/nexus
qmca H2O.s002.scalar.dat # For statistical analysis of the DMC data
qmca -t -q e H2O.s002.scalar.dat # Graphical plot of DMC energy
The last command will produce a graph as per Fig. 1. This shows the average energy of the DMC walkers at each timestep. In a real simulation we would have to check equilibration, convergence with walker population, time step, etc.
Congratulations, you have completed a DMC calculation with QMCPACK!
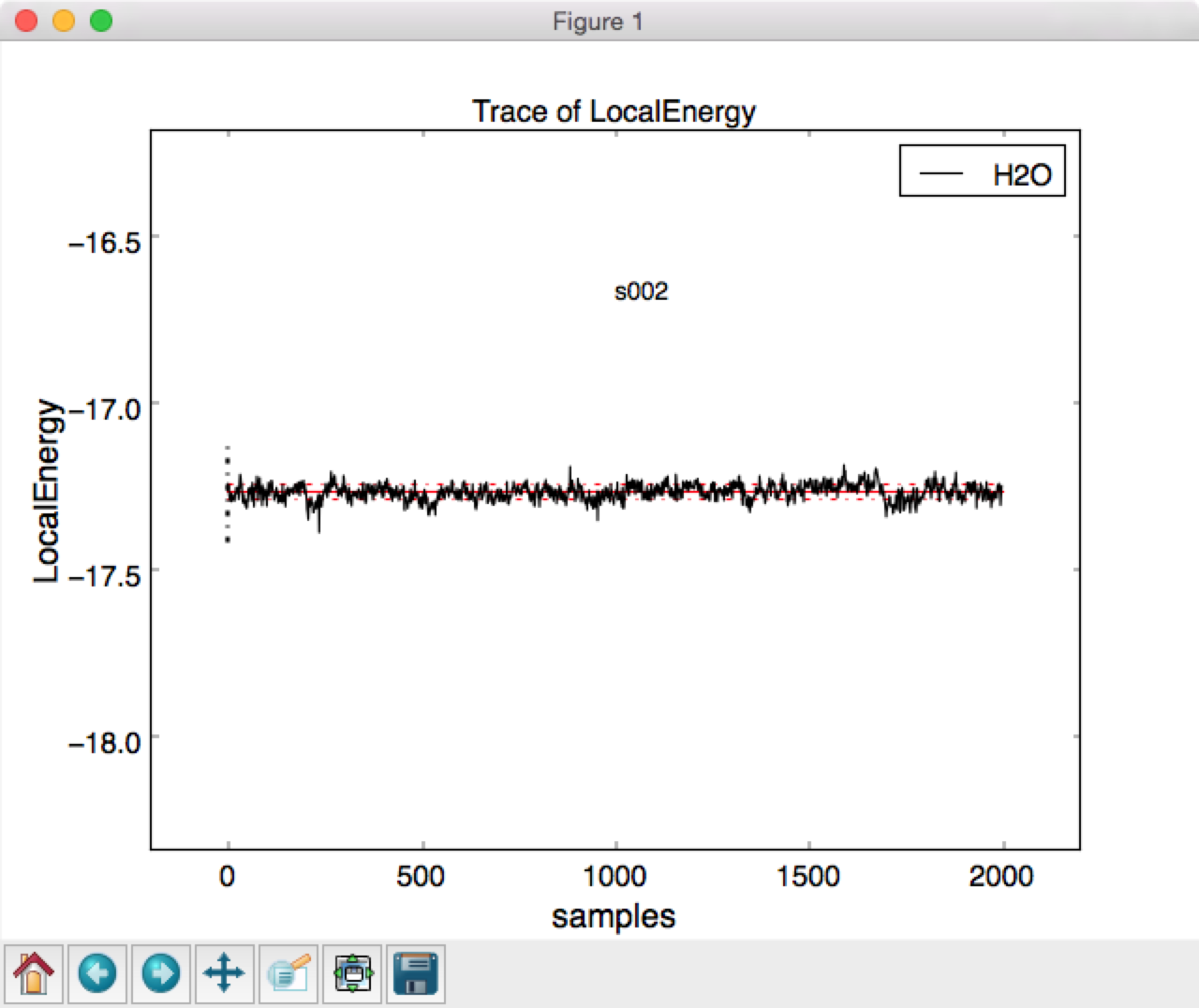
Fig. 1 Trace of walker energies produced by the qmca tool for a simple water molecule example.
Support and Contacting the Developers
Questions about installing, applying, or extending QMCPACK can be posted on the QMCPACK Google group at https://groups.google.com/forum/#!forum/qmcpack or as an issue on the QMCPACK GitHub repository https://github.com/QMCPACK/qmcpack/issues. You may also email any of the developers, but we recommend checking the group first. Particular attention is given to any problem reports.
Performance
QMCPACK implements modern Monte Carlo (MC) algorithms, is highly parallel, and is written using very efficient code for high per-CPU, GPU or on-node performance. In particular, the code is highly vectorizable, giving high performance on modern central processing units (CPUs) and graphics processing units (GPUs). We believe QMCPACK delivers performance either comparable to or better than other QMC codes when similar calculations are run, particularly for the most common QMC methods and for large systems. If you find a calculation where this is not the case, or you simply find performance slower than expected, please post on the Google group or contact one of the developers. These reports are valuable. If your calculation is sufficiently mainstream we will optimize QMCPACK to improve the performance.
Open Source License
QMCPACK is distributed under the University of Illinois at Urbana-Champaign/National Center for Supercomputing Applications (UIUC/NCSA) Open Source License.
University of Illinois/NCSA Open Source License
Copyright (c) 2003, University of Illinois Board of Trustees.
All rights reserved.
Developed by:
Jeongnim Kim
Condensed Matter Physics,
National Center for Supercomputing Applications, University of Illinois
Materials computation Center, University of Illinois
http://www.mcc.uiuc.edu/qmc/
Permission is hereby granted, free of charge, to any person obtaining a
copy of this software and associated documentation files (the
``Software''), to deal with the Software without restriction, including
without limitation the rights to use, copy, modify, merge, publish,
distribute, sublicense, and/or sell copies of the Software, and to
permit persons to whom the Software is furnished to do so, subject to
the following conditions:
* Redistributions of source code must retain the above copyright
notice, this list of conditions and the following disclaimers.
* Redistributions in binary form must reproduce the above copyright
notice, this list of conditions and the following disclaimers in
the documentation and/or other materials provided with the
distribution.
* Neither the names of the NCSA, the MCC, the University of Illinois,
nor the names of its contributors may be used to endorse or promote
products derived from this Software without specific prior written
permission.
THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS
OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY,
FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL
THE CONTRIBUTORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR
OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE,
ARISING FROM, OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR
OTHER DEALINGS WITH THE SOFTWARE.
Copyright is generally believed to remain with the authors of the individual sections of code. See the various notations in the source code as well as the code history.
Contributing to QMCPACK
QMCPACK is fully open source, and we welcome contributions. If you are planning a development, early discussions are encouraged. Please post on the QMCPACK Google group, on the QMCPACK GitHub repository, or contact one of the developers. We can tell you whether anyone else is working on a similar feature or whether any related work has been done in the past. Credit for your contribution can be obtained, for example, through citation of a paper or by becoming one of the authors on the next version of the standard QMCPACK reference citation.
See Development Guide for details about developing for QMCPACK, including instructions on how to work with GitHub, the style guide, and examples about the code architecture.
Contributions are made under the same license as QMCPACK, the UIUC/NCSA open source license. If this is problematic, please discuss with a developer.
Please note the following guidelines for contributions:
Additions should be fully synchronized with the latest release version and the latest develop branch on GitHub. Merging of code developed on older versions is error prone.
Code should be cleanly formatted, commented, portable, and accessible to other programmers. That is, if you need to use any clever tricks, add a comment to note this, why the trick is needed, how it works, etc. Although we appreciate high performance, ease of maintenance and accessibility are also considerations.
Comment your code. You are not only writing it for the compiler for also for other humans! (We know this is a repeat of the previous point, but it is important enough to repeat.)
Write a brief description of the method, algorithms, and inputs and outputs suitable for inclusion in this manual.
Develop tests that exercise the functionality that can be used for validation and for examples. Where is it practical to write them, we prefer unit tests and fully deterministic tests ahead of stochastic tests. Stochastic tests naturally fail on occasion, which is a property that does not scale to hundreds of tests. We can help with this and tests integration into the test system.
QMCPACK Roadmap
A general outline of the QMCPACK roadmap is given in the following sections. Suggestions for improvements from current and potential users are very welcome, particularly those that would facilitate new uses or new users. For example, if an interface to a particular quantum chemical or density functional code, or an improved tutorial would be helpful, these would be given strong consideration.
Code
The codebase is being transitioned to a single performance portable design, often referred to as the batched code. As of the beginning of 2025, these codepaths are now the default, have been heavily tested, and also used for numerous publications. The batched codepaths largely offer the same feature set on all platforms, whether CPU systems or GPUs from NVIDIA, Intel, or AMD. Unlike QMCPACK’s original GPU implementation, the intent of the new code is for calculations to fallback to CPU execution where GPU implementations are not available. The implementations will be optimized and further matured based on the feedback received applying them to current science problems.
We will continue to improve the usability and reliability of QMCPACK through combinations of more convenient input parameters, improved workflows, integration with more quantum chemical and density functional codes, and a wider range of examples. Suggestions are very welcome, both from new users of QMC and from those experienced with other QMC codes.
Documentation and examples
This manual describes the core features of QMCPACK that are required for routine research calculations and standard QMC workflows, i.e., the VMC and DMC methods, auxiliary field QMC, how to obtain and optimize trial wavefunctions, and simple observables. This covers at least 95% of use cases, and nearly all production research calculations.
Because of its history as an academically developed research code, QMCPACK also contains a variety of additional QMC methods, trial wavefunction forms, potentials, etc., that while not mature, might be useful for specialized calculations on particular material or chemical or model systems. If you are interested in these please ask – we might have historical inputs available. For example, if an older/historical Jastrow factor form that was previously used for a single paper is needed for benchmarking, we can look into the status.
P. R. C. Kent, Abdulgani Annaberdiyev, Anouar Benali, M. Chandler Bennett, Edgar Josué Landinez Borda, Peter Doak, Hongxia Hao, Kenneth D. Jordan, Jaron T. Krogel, Ilkka Kylänpää, Joonho Lee, Ye Luo, Fionn D. Malone, Cody A. Melton, Lubos Mitas, Miguel A. Morales, Eric Neuscamman, Fernando A. Reboredo, Brenda Rubenstein, Kayahan Saritas, Shiv Upadhyay, Guangming Wang, Shuai Zhang, and Luning Zhao. QMCPACK: Advances in the development, efficiency, and application of auxiliary field and real-space variational and diffusion quantum Monte Carlo. Journal of Chemical Physics, 152(17):174105, May 2020. doi:10.1063/5.0004860.
Jeongnim Kim, Andrew T. Baczewski, Todd D. Beaudet, Anouar Benali, M. Chandler Bennett, Mark A. Berrill, Nick S. Blunt, Edgar Josué Landinez Borda, Michele Casula, David M. Ceperley, Simone Chiesa, Bryan K. Clark, Raymond C. Clay III, Kris T. Delaney, Mark Dewing, Kenneth P. Esler, Hongxia Hao, Olle Heinonen, Paul R. C. Kent, Jaron T. Krogel, Ilkka Kylänpää, Ying Wai Li, M. Graham Lopez, Ye Luo, Fionn D. Malone, Richard M. Martin, Amrita Mathuriya, Jeremy McMinis, Cody A. Melton, Lubos Mitas, Miguel A. Morales, Eric Neuscamman, William D. Parker, Sergio D. Pineda Flores, Nichols A. Romero, Brenda M. Rubenstein, Jacqueline A. R. Shea, Hyeondeok Shin, Luke Shulenburger, Andreas F. Tillack, Joshua P. Townsend, Norm M. Tubman, Brett Van Der Goetz, Jordan E. Vincent, D. ChangMo Yang, Yubo Yang, Shuai Zhang, and Luning Zhao. QMCPACK : an open source ab initio quantum Monte Carlo package for the electronic structure of atoms, molecules and solids. Journal of Physics: Condensed Matter, 30(19):195901, 2018. doi:10.1088/1361-648X/aab9c3.